
Atrézie jícnu
Atrézie jícnu je vrozená vada, při které je jícen nesprávně vyvinutý. Většinou je slepě ukončen a často bývá spojen píštělí s tracheou (až v 85%). Vyskytuje se u jednoho z 2000 – 4000 novorozenců, dívky i chlapci bývají postiženi stejně často. Tato vada se projevuje ihned po porodu, novorozenec má pěnivý sekret v dutině ústní, při prvním pití kašel, cyanóza, aspirace při krmení a potíže s dýcháním. Atrézie jícnu je závažný stav, který vyžaduje okamžitou lékařskou péči (po porodu se vždy dává sonda do jícnu – resp. jeho horního slepého vaku, miminko je ve zvýšené poloze) a překládá se na specializované pracoviště k chirurgickému řešení. Co nejdříve se musí uzavřít píštěl kvůli nebezpečí aspirace. Je-li to možné, tak horní a dolní konec se sešijí tzv. end-to end. Pokud k sobě nedosáhnou, hledají se další možnosti řešení.
Typ H píštěle, kdy není jícen přerušen, jen je přítomna píštěl s průdušnicí, se může projevit i později po porodu.
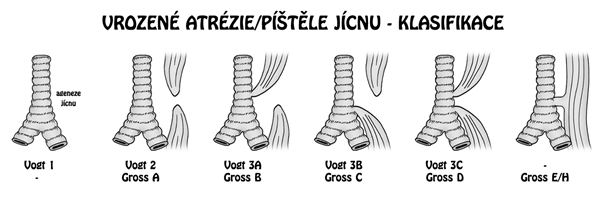
Atrézie jícnu má několik typů, které se klasifikují dle Vogta nebo Grosse:
- typ I: úplné chybění jícnu nebo místo jícnu vazivový pruh, <1 %
- typ II: dva vzdálené pahýly, píštěl nepřítomna, 8 %
- typ IIIa: horní ezofagotracheální píštěl, dolní slepý vak 1 %
- typ IIIb: dolní ezofagotracheální píštěl, horní slepý vak, nejčastější (85 – 90 %)
- typ IIIc: horní a dolní ezofagotracheální píštěl 1 %
- H-píštěl: jícen průchodný, přítomna píštěl ve tvaru H mezi jícnem a tracheou 5 %
Atrézie a stenózy tenkého a tlustého střeva
Atrézie a stenózy tenkého a tlustého střeva jsou vrozené vady, které ovlivňují funkci trávicího systému. Vyskytují se u jednoho z 3 000 – 5 000 novorozenců. Atrézie je stav, kdy je část střeva úplně uzavřená nebo chybí, zatímco stenóza je zúžení střeva, které může omezovat průtok potravy a tekutin. Tato postižení mohou být i mnohočetná a střevo bývá postiženo v různé délce.
Klinika se liší dle lokalizace a tíže postižení, symptomy se obvykle projeví krátce po narození. Nejčastější je napětí břišní stěny, bříško je vzedmuté, zvracení, poruchy vyprazdňování stolice (první smolka ale běžně odchází), dále nestabilní teplota či dehydratace.
Léčba atrézie a stenózy tenkého a tlustého střeva je chirurgická, kdy je obnovena kontinuita střev. Přežití a dlouhodobé výsledky se liší a závisí na závažnosti stavu, místě postižení ve střevě a celkovém zdraví dítěte.
Pylorostenóza
Pylorostenóza neboli hypertrofická stenóza pyloru není vrozenou vadou, zde je zmíněna jen pro úplnost. Jedná se o získané ztluštění hladké svaloviny pyloru, což je část mezi žaludkem a tenkým střevem. Příčiny nejsou známé, ale až v 15 % je prokázaný rodinný výskyt. Vyskytuje se přibližně u jednoho z 5 000 novorozenců, až 5x častěji u chlapců. Onemocnění se nejčastěji projeví mezi 3 – 6. týdnem po porodu. Klinicky dominuje explozivní zvracení obloukem (až do 1 m), dle tíže zvracení se pak přidává dehydratace s metabolickým rozvratem. Léčba je chirurgická.
Atrézie žlučových cest
Atrézie žlučových cest je závažná vrozená vada, při které dochází k blokádě nebo k úplnému chybění žlučových cest, které umožňují žluči cestovat z jater do střeva. Mohou být postiženy jak úseky v játrech (intrahepatické žlučové cesty), tak části mimo játra (extrahepatické žlučové cesty). Tento stav vede většinou k vážnému poškození jater s následným selháním a stává se tak nejčastějším důvodem pro transplantaci jater u dětí. Tato vada se vyskytuje u jednoho u 8 000 – 20 000 novorozenců (u Asiatů významně častěji než u Evropanů).
Symptomy atrézie žlučových cest se obvykle objevují v prvních dvou měsících života. Po počátečním ústupu novorozenecké žloutenky dochází k opětovnému nárůstu bilirubinu, což se projeví znovu žloutenkou, kůže je zbarvena do žlutozelena. Moč je zbarvená bilirubinem do tmava, stolice bez bilirubinu je naopak světlá, zvětšují se játra. I pokud je atrézie žlučových cest včas diagnostikována a léčena, může vést k cirhóze jater a selhání jater.
Léčba atrézie žlučových cest je chirurgická, nejčastěji se používá operace tzv. portoenteroanastomóza dle Kasaie, která se snaží obnovit odtok žluče z jater do střev.
Omphalokéla
Omphalokéla je vlastně druh vrozené pupeční kýly, kdy se obsah dutiny břišní vyklenuje do pupečníku (nejčastěji střevo). Pupečník nasedá na vrchol kýlního vaku. S touto vadou se rodí asi jedno z 5 000 dětí. Velikost omphalokély se může lišit, v některých případech může obsahovat pouze malou část střev, v jiných případech může obsahovat i velkou část břišních orgánů.
Omphalokéla je viditelná již prenatálně při ultrazvukovém vyšetření a i okamžitě po narození. Tento stav může být spojen s dalšími vrozenými vadami, jako jsou srdeční vady, defekty střev a genetické poruchy. Léčba omphalokély je chirurgická. Děti s touto vadou se rodí plánovaným císařským řezem na specializovaném pracovišti.
Laparoschíza
Laparoschíza neboli gastroschíza je vrozená vada (cca 1:5 000 novorozenců), při které se bříško neuzavřelo ve svém vývoji. Otvor v břišní stěně dítěte je obvykle vpravo od pupeční šňůry. Tímto otvorem vyhřezávají střeva a někdy i jiné orgány, jako jsou játra a žaludek, ven z těla. Tato vada je vidět také již na prenatálním ultrazvukovém vyšetření a samozřejmě je viditelná ihned po porodu. Na rozdíl od omphalokély, orgány nejsou ničím obalené a jsou přímo vystaveny plodové vodě, což vždy způsobí peritonitidu (zánět). Děti s laparoschízou se rodí plánovaným císařským řezem na specializovaném pracovišti. Léčba laparoschízy obvykle zahrnuje několik operací, při kterých jsou orgány opatrně vráceny zpět do těla a břišní stěna je uzavřena.